HE26
 HE313.6548882425E+14 72
HE313.6548882425E+14 72
0
All programs expect an atomic data file, which contains model atoms and atomic data. It is firstly created by the user (Sect. 2.7) and then processed by the program ATOMS2.
The model ions of one element have to be inserted in increasing order while any order of elements is accepted.
The processing of the atomic data file is directed by keywords which meanings are explained in the following. All keywords as well as all strings have to be inserted flush left (e.g. level names, Sect. 2.1). All numerical data can be written format free. Attention: all input lines have a maximum length of 80 characters!
The levels are named using an A10 string. The element code is at the beginning (e.g. HE) and the following (one or two) numbers indicate the ionization stage (e.g. HE2). This is mandatory. The next numbers indicate the principal quantum number of the level (e.g. HE26). For levels with a complicate configuration, the electron system and the magnetic quantum number may be added (e.g. O42P 2PO). Singly excited levels are marked by a “ ’ ”, doubly excited by a “ ’ ’ ” directly before the electron system indication (e.g. N33S’ 2PO). In the case of explicitly considered multiplet splitting the angular momentum is added at the end (e.g. C452D5/2).
.
0
ATOM
L
LTE
RBB
RBF
RDI
RLL
RLU
CBB
CBF
CBX
TL
DB
The expression “card” which is frequently used in the following is a relic from the PUNCH era and is used for historical reasons.
keyword .
comment, not written to atomic data file
keyword 0 (Null)
necessary to end the validity of some keywords (e.g. L, LTE, ...)
keyword ATOM
introduces a new element. The following card indicates:
Example:
ATOM
HE 0 4.0026
means: all following cards describe an helium model atom, starting with neutral helium He I, the atomic weight is 4.0026 AMU.
keyword L
introduces non-LTE levels. All cards following this keyword indicate:
Example:
L
HE26HE313.6548882425E+14 72
0
levels of an ionization stage are expected in increasing energetic order (from the ground state). The keyword 0 completes the list of NLTE levels for this ionization stage.
keyword LTE
introduces LTE levels analogously to the keyword L. Do not forget to complete the list with the keyword 0. Important: some formulae for the free-free opacity (Sect. A) expect at least one LTE level in the respective ion. Attention: this is not checked by the program.
Radiative and collisional transitions are introduced by:
All lists following these keywords have to be completed by the keyword 0. A card which follows one of the keywords (except RDI) indicates
Example:
RBB
H11H131 1 0.0791
0
This is the line transition Ly , cross-section calculation with formula No. 1, one input number
(0.0791).
, cross-section calculation with formula No. 1, one input number
(0.0791).
For RDI transitions a third level is introduced between lower and upper level which is the upper level of the stabilizing transition and has to be introduced before as a LTE level.
For RLU and RLL transitions the level names are followed by the name of the file which contains the “sample” cross-section.
Example:
RLU
FE31FE3326_02_01_03
0
keyword RFF
introduces radiative free-free transitions. The following card indicates:
keyword TL
The card which follows this keyword indicates the “line temperature” Tline for all following RBB
transitions. This is necessary to change the temperature used for the calculation of the Doppler width
(default Tline = 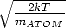 Teff) in the creation of the frequency grid (Sect. 3). The default is again valid
after
Teff) in the creation of the frequency grid (Sect. 3). The default is again valid
after
TL
0
keyword DB
introduces an explicit frequency grid for all following RBB transitions. The following card indicate:
Example:
DB
7
-3 -2 -1 0 1 2 3
If the first frequency point is 0.0, only “half” lines are created.
This grid is valid until the next keyword DB appears. The default is valid after
DB
0
Note: In the Tübingen Model-Atom Database (Sect. 2.4), standard DB values are given
that are about the maximum needed at low effective temperatures and high surface
gravities. These values have to be altered at least for lower surface gravities. A test
calculation with PRO2 will show the line widths. Use, e.g., H i 1215 Å, and reduce the
maximum DB values. In case that they are too low, “steps” in the flux level are
prominent in the line wings. (The Doppler width of a specific line can be calculated by
/home/rauch/bimod/doppler.Linux_x64.)
1215 Å, and reduce the
maximum DB values. In case that they are too low, “steps” in the flux level are
prominent in the line wings. (The Doppler width of a specific line can be calculated by
/home/rauch/bimod/doppler.Linux_x64.)
The DB adjustment results in a lower number of frequency points and, thus, faster model calculation.
A good compromise may also be to replace
| 1cAp0x9-900012: |
by the insertion of a grid of additional frequency points using, e.g.,
and a reduced DB that represents the line core much finer than the base grid (for F_BASE see Sect. 3)
The Tübingen model-atom database TMAD ( http://astro.uni-_tuebingen.de/~TMAD) provides ready-to-use model atoms in TMAP format.
TMAD provides, in general, model atoms at maximum size, i.e. using all data from standard atomic-data resources. For model calculations, model ions can be reduced typically to 15 to 20 NLTE levels (Sect. 2.5, cf. Jahn et al. 2007) to avoid unreasonable computation times. In addition, the input for the DB keyword (Sect. 2.5) has to be adjusted to avoid calculations of line profile very far off the line centers where the line absorption is negligible. TMAD uses a standard of 2400 Doppler widths which may be necessary only for models with very high surface gravities.
Models from Sect. 2.4.1 may not include all levels, e.g. for C iv lines in the optical wavelength range around 4660 Å. Thus, a subsequent line-formation calculation has to be performed with fixed atmospheric structure, i.e. only NLTE occupation numbers for the atomic levels are calculated. It may not be useful to extend all ions and to consider all levels given in TMAD. The cookbook says, “for all ions for which lines are identified, include five more levels than the highest from which strategic lines are arising”.
In case of the C iv 4660 Å / He ii 4685 Å absorption trough in PG 1159 stars, it is sufficient to
extend C iv only for a line-formation calculation, even in models that consider all species from H to
Ni.
LINE1 and LINE1_PROF provide the possibility to collect occupations numbers of newly considered levels from different models Sect. 8.3, e.g. if five line-formation calculations were performed (based on the same model) with individually extended C iv, O v, O vi, Ne vi, and Ar vi. This saves an enormous amount of CPU time.
To calculate the emergent synthetic spectrum, a formal solution is performed with LINE1_PROF. TMAD provides model atoms that account for fine-structure splitting of the atomic levels. These have to be the same like the unsplitted levels used for the line-formation calculations. (Sect.2.4.2).
The interactively created atomic data file is — if designed following the instructions in Sect. 2 — ready, i.e. it can be used for the creation of a frequency grid as well as for model atmosphere calculations. However, it is highly recommended to process it with the program ATOMS2, which is able to detect a lot of errors and gives warnings. In case of fatal errors it even terminates. There exist options which make ATOMS2 create automatically model ions or fill up them.
The program ATOMS2 is available at Tübingen’s PC/Workstation cluster in its latest version:
/home/rauch/bimod/atoms2.Linux_x64.
An atomic data file (ATOMS) created by ATOMS2 is unambiguously defined by:
In the input file OPTIONEN one can write directives for ATOMS2. ATOMS2 expects
OPTIONEN and ATOMIN subsequently as input, OPTIONEN has to be finished in any case
with
ENDOPTIONS,
even if none of the following options is given:
AUTOIONHI in il ir
This option creates a complete H model atom. in is the principal quantum number of the
highest H I NLTE level, il is the principal quantum number of the highest H I LTE level,
ir is the principal quantum number of the highest H I NLTE levels up to which all RBB
transitions are considered. (The conditions il > in and ir < in are fulfilled.) In case that
ir is negative, only transitions from ATOMIN up to the NLTE level ir are used. (This is
necessary if ATOMIN contains information about line broadening, default is Doppler line
broadening). In the case that ir is positive, ATOMIN does not need to contain any H model
atom.
AUTOIONHEII in il ir
same like AUTOIONHI (see above) but mandatory; in the case that ATOMIN contains a He I
model ion, ATOMS2 creates He II and He III model ions, otherwise a complete He model
atom.
CBB-AUTO-FILL
If this option is given, missing CBB transitions are automatically inserted and possible errors in
ATOMIN corrected — as far as ATOMS2 is able to detect them. The output of ATOMS2 has to be
checked for respective error messages or warnings.
CBB-AUTO-FILL(IGNORE)
same like CBB-AUTO-FILL; possible errors are only reported in the output but not corrected.
CBB-AUTO-FILL(NONE)
If this option is given, all CBB transitions are disregarded. This ...-AUTO-FILL option is only useful for
the detailed calculation line profiles. This option is automatically set, if the option LINEFORMATION ... is
given (see below).
Analogously to the CBB-AUTO-FILL ... options, the following options are valid:
CBF-AUTO-FILL
(for unknown cross-sections, hydrogen-like values are automatically inserted)
CBF-AUTO-FILL(NONE)
CBF-AUTO-FILL(NOOP)
(for OpacityProject cross-sections, hydrogen-like values are automatically inserted)
CBX-AUTO-FILL
CBX-AUTO-FILL(IGNORE)
CBX-AUTO-FILL(NONE)
RBB-AUTO-FILL(NONE)
RBB-AUTO-FILL(H1)
RBB-AUTO-FILL(HE2)
RBF-AUTO-FILL
RBF-AUTO-FILL(NOOP)
(for OpacityProject cross-sections, hydrogen-like values are automatically inserted)
(set CBF-AUTO-FILL(NOOP) also)
RDI-AUTO-FILL(NONE)
RLL-AUTO-FILL(NONE)
RLU-AUTO-FILL(NONE)
LINEFORMATION-RBB-INTERVALL=[1000,7000],ION=H1
This option can be set for line-profile calculations in order to restrict number of line transitions in the
atomic data file ATOMS to a selected interval (here: [1000, 7000] Å). If this shall be valid for all (line
profiles), ION=NON has to be inserted. To insert all line transitions — i.e. even outside the given interval
— of a selected ion in ATOMS (line formation), the TMAP code (Sect. 2.1, here: H1) has to be
given.
If this option is given, all
C.. -AUTO-ION(NONE)
options and the
RDI-AUTO-ION(NONE)
option are automatically set.
LISTOFALLRBB,ION:
This option creates a table of all line transitions of the selected ion under consideration of a possible
LINEFORMATION ... option with its interval restriction.
ENDOPTIONS
This option finishes the file OPTIONEN and is necessary if one of the other options has been given.
The program ATOMS2 creates a large informative output which includes generally the atomic data file as created by ATOMS2, informations about wavelengths of levels, thresholds, line transitions, and some statistics. Excited levels are marked in the output with “**”, all transitions which were inserted following the ...AUTO-FILL ... options are marked with “AF”.
All parameters which are valid for the created atomic data file ATOMS (Sect. 1), are summarized in a table at the end of the output. Under unix a
grep para ¡output filename¿
extracts them from the output file.
The Tübingen Iron-Group Opacity (TIRO) service creates atomic data files and cross-section data for radiative bound-bound and bound-free transitions of iron-group elements (calcium, scandium, titanium, vanadium, chromium, manganese, iron, cobalt, and nickel). It is based on the program IrOnIc that was developed at Tübingen. TIRO enables the VO user to consider iron-group elements in model-atmosphere calculations easily, in various ways, and without spending own calculation time for the creation of the necessary input data. It is controlled via web interface ( http://astro.uni-_tuebingen.de/~TIRO) in which the following inputs have to be given.
After submitting the data, the given parameters are stored in a request file. TIRO checks regularly if requests are waiting and processes them one after the other. The user is informed via email when the handling of the data starts. The resulting files are stored in a compressed tar archive that is accessible via a wget command. The user is informed via email about its location. The files for bound-bound transitions contain a table with frequencies in the first column, cross-sections in the second (calculated for electron density 0) and in the third column (calculated for electron density 1016/cm3). The corresponding files for bound-free transitions include a table with frequencies in the first and cross-sections in the second column.
Some programs are currently available which help to design model atoms etc. All these programs work interactive and are (more or less :-( ...) self-explaining.
The programs PRO2 and LINE1 can consider – besides pure Doppler line broadening – the quadratic (“formula 3”) and the linear (“formula 4” — this gives the name of the program ...) Stark effect for the line broadening. Both formulas need some input data which has to be includes in the atomic data file ATOMS. (Sect. 2).
The program FORMEL4 (/home/rauch/bin/formel4) calculates these data (mainly the classical damping constant and the effective quantum number) from an existing atomic data file.
The calculated data is saved into the files FORMEL4.DAT and FORMEL4.RBB. FORMEL4.RBB can be used the replace the respective section of the atomic data file which was used to calculate the data.
FORMEL4 inserts also the vacuum wavelengths, starting with the keyword WAVELENGTH: in columns 81-91. The wavelength values may be the replaced by measured wavelengths in order to shift those lines which arise from levels with uncertain energies to the correct wavelengths. A subsequent run (necessary!) of SETF2 on the modified ATOMS yield a frequency grid with these lines at the correct wavelengths.
The level energies in literature are commonly given in cm-1, measured from the ground state of their ionization state. In the Tübingen NLTE Model-Atmosphere Package (TMAP), the level energies given are the difference to the next ionization limit, i.e. the energy which is necessary to ionize into the ground state of the next ionization stage. The program LEVEL simply transforms these energies.
The program LEVEL (/home/rauch/bin/level) runs interactively. It is possible to calculate
• energies of single levels
• energies of combined levels
Furthermore it is possible to combine
• levels from an existing atomic data file
The data is saved into LEVEL.DAT and (for the combination of levels) into XX.ENTARTET (XX is the principal quantum number of the combined levels).
Due to our statistical approach to create atomic data files from Kurucz’s data (Kurucz 1991, 2009, 2011) of
elements with atomic numbers Z > 20, the wavelengths of individual lines can only be read from
Kurucz’s data files. The two procedures
/home/rauch/tools/lineidentification_LIN.bat and
/home/rauch/tools/lineidentification_POS.bat
for Kurucz’s data files with theoretical and laboratory measured lines (.LIN) and with positively
identified lines (.POS), respectively) may be used to create
\IDENT <wavlength> <ion>
cards that can be used by WRPLOT (Sect. 11). An explanation how to use these procedures is found in
their headers.
During the calculation of the model atmospheres, multiplets are considered with a combined level. For the subsequent detailed line profile calculation, it is necessary to split these multiplets into single components. While (in most cases) the level energies can be taken from literature, there are only rudimentary informations about the oscillator strengths of the single components. The program MULTIPLET (/home/rauch/bin/multiplet) splits up oscillator strengths of doublets, triplets, and quartets under the assumption of LS coupling.
The calculated data is saved into MULTIPLET.DAT.
For some RBF cross-sections of levels of the ions He I, C I – C IV, N I – N V, O I – O III, Ne I – Ne II, Mg I – Mg II, Si I – Si IV etc., tables have been calculated by Hofs”a”s. These can be evaluated with the program SEATON (). One gets input parameter for the Seaton formula(Sect. A.4). For those levels which are not included in the tables, SEATON can calculate a hydrogen-like cross-section.
To reduce the pixel-to-pixel variation (or the noise) of observations, a low-pass filter may be used. The program SGF (/home/rauch/bin/sgf) processes data following Savitzky & Golay (1964). Two parameters, np and m have to be given. np is the number of a subset of the data which is used for a fit. m is the degree of the polynomial used in the least-squares fit method. m has to be carefully chosen - a too-high value smoothes out real spectral features.